| 1. 溶媒和構造の変化を通じた化学反応エネルギーの力学的仕事への変換メカニズムの解明 |
| |
生体内では、筋肉中のアクチン-ミオシン系等の様に化学反応エネルギー(例えばATPの加水分解反応)
を力学的仕事として利用する系の存在が多数知られています。
言うまでもなく生体内の現象は溶液内(凝縮系)での現象であり、
溶媒分子を含めた莫大な自由度の関与している系です。
そうした凝縮系での化学反応の力学的仕事への変換機構はいくつか考えられます。
本研究テーマでは、化学反応によってモーター分子の表面状態の変化が溶媒和状態の変化を引き起こし、
その変化がモーター分子に対する仕事に繋がる場合を考えます。
ここでは、こうした機構によるモーターを溶媒和モーター(Solvation Motor、SM)と呼ぶことにします。
溶媒分子を露に扱う分子動力学(MD)シミュレーションを行い、
このSM機構の可能性を確認し、その非平衡挙動を分子論的に捉えることを目標としています。
論文投稿できる程度に結果がまとまったのち、内容の詳細を掲載します。
|
| |
| |
| 2. 分子型量子ドットセルオートマトン(分子QCA)の動作解析&理論設計 |
| |
| 1.量子ドットセルオートマトン(QCA)とは? |
|
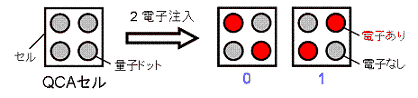
まず、4つの量子ドットを1組にして、「セル」を作ります。
次に、セルに2つの電子を注入すると、2個の電子はクーロン力により反発するため、
下図のように2つの縮退した安定状態が生じることが分かります。
これらを、「0」, 「1」と定義すれば、このセルはデジタルの情報を
を表わしているということができます。
このように、量子ドットを組み合わせて、0と1のデジタル情報を表すものを
量子セルオートマトン(QCA)と呼びます。
|
 |
| |
| 2.QCAで作る論理回路 |
|
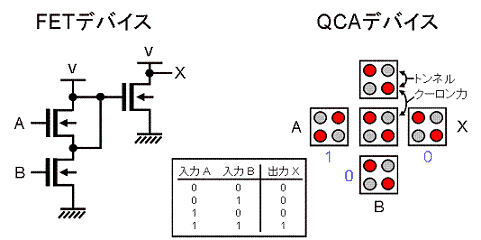
コンピュータの中央演算装置(CPU)は、ANDやORといった論理回路から作られています。
さらに、論理回路は電界効果トランジスター(FET)を組み合わせることによって作られています。
下図・左にFETから構築したAND回路の例を示しています。
実は、同じようなAND回路は、上述のQCAセルを組み合わせることによって作成することもできます(下図・右)。
そして、2000年前後に、このQCAデバイスが論理回路として動作することが実験的に示されています。
|
 |
| |
| 3.「QCA」から「分子QCA」へ・・・ |
QCAデバイスが論理回路として動作することは既に実証されていますが、
・デバイス動作が、量子ドットのサイズ・位置に大きく依存する
・常温では、0と1がランダムに入れ替わるため、正常に動作しない
・セル間のクーロン相互作用が弱いため、信号伝達強度が弱い
といった問題点が指摘されています。
これらの問題点を解決するために、QCAセルを1分子で代用する「分子QCA」
のアイデアが提唱されました。
理論上は、分子QCAは上述の問題点を解決できると考えられていますが、
まだ分子QCAを用いたデバイスの実験的な動作は実証されていません。
現在、分子QCAの実用化を目指して、
・分子QCAの演算挙動のシミュレーション
・分子QCAとして有用な分子の理論的提唱
を目的に研究を行っています(論文 12, 17、本・雑誌 6, 7)。
分子QCAデバイスの原理、および、これまでの流れに関しては、雑誌 6 をご覧下さい。
|
| |
| 3.進捗状況 |
まず、Creutz-Taube錯体(ルテニウム2核錯体)を分子QCAの例として取りあげ、
QCA内の信号伝達挙動の評価・解析方法の確立を行った(論文 12)。
錯体の近くに点電荷を配置し、その電荷の変化をQCAに対するスイッチと考えた。
信号伝達挙動の簡単な描像を得るため、密度汎関数法(DFT)およびHartree-Fock(HF)法
といった1電子近似に基づく方法を用いた。
シミュレーションの結果、スイッチに伴い錯体内を伝達する信号の強度(電子の量)は、
点電荷によるHOMO, LUMOの軌道分布の歪みに密接に関係していることが分かった。
また、スイッチに伴い錯体内を伝達する信号の速度は、
HOMO-LUMOギャップの大きさに反比例することが分かった。
次に、Creutz-Taube錯体を取りあげ、金属原子として Fe, Ru, Os を選び、
分子QCA内の信号伝達挙動の金属原子依存性を解析した(論文 17)。
以上の2報の内容は、フリーアクセスの本・雑誌 7 にまとめてあります。
|
| |
| |
| 3. キャリア輸送材料の量子化学的設計 |
| |
半導体層に有機半導体材料を用いる有機半導体デバイスは、シリコンを用いる無機半導体デバイスに代わるものとして期待されています。
なぜなら、有機半導体材料には、「柔軟性」や「軽量性」といった大きな利点があるためです。
それに加えて、有機半導体材料は、化学的な方法でその物性を様々にチューニングできる「多様性」という特徴を有しています。
半導体材料は2つに分けることができます。
一つは電子をキャリアとする電子輸送材料(n-チャンネル半導体)、
もう一つはホール(正孔)をキャリアとするホール輸送材料(p-チャンネル半導体)です。
半導体デバイスの性能を支配する因子は幾つかありますが、重要なものの1つが半導体材料中の電子・ホールの「移動度」です。
「キャリアの移動速度」=「キャリアの移動度」×「電場の強さ」の関係があるため、
移動度の大きい材料ほど、スイッチに対するデバイスの応答が素早いと言えます。
ゆえに、一般的に、この移動度の大きい材料が半導体材料として優れています。
このため、有機半導体材料にも、従来の無機半導体材料に匹敵する移動度(約 1 cm2V-1s-1)を有することが求められてきました。
我々(徳永、大森、川畑)は、フラーレンC60誘導体の有機半導体材料としての可能性に注目して、研究を進めています。
なぜC60そのものではなく、C60誘導体なのか?
実は、C60誘導体を研究している理由は、電子輸送材料とホール輸送材料で異なります。
それぞれについて、その理由と成果の一部を以下に示しています。
|
| |
| 1.C60誘導体のホール輸送材料(p-チャンネル半導体)としての利用 |
C60材料のホール移動度は10-4cm2V-1s-1オーダーであると報告されています。
このため、C60そのもののホール輸送材料としての価値は低いと言えます。
そこで、我々は、C60をC60誘導体とすることにより、
ホール移動度を増加させることができるのではないかと考えました。
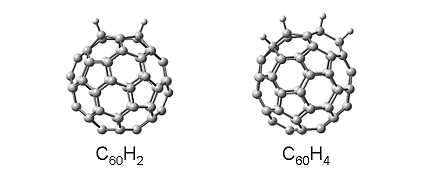
現在、最も単純なC60誘導体である水素化フラーレン:C60Hnに関して研究を進めています。
これまでに、ホール移動度の値はC60の値と比較して
C60H2で約2.5倍(論文 9、雑誌 2)、
C60H4で約3.3倍(論文 10、雑誌 2)、
に増加することを報告しています。
|
 |
また、このように水素化によりホール移動度が増加する理由として、
・水素化により、カチオン状態での電子状態の縮退が解ける
・注入されたキャリアの一部が水素原子付近に局在する
ことにより、再配列エネルギーが減少することを提唱しています(論文 9, 10、本・雑誌 2)。
さらに、C60H1、C60H6、
についての考察を行うとともに(論文 20)、
C70フラーレン誘導体(C70H1、C70H2)へと対象を拡げています
(論文 18, 20)。
得られた知見に基づき、さらに大きなホール移動度を有するフラーレン誘導体材料の設計を目指します。
|
| 2.C60誘導体の電子輸送材料(n-チャンネル半導体)としての利用 |
C60材料の電子移動度は、
真空蒸着により作成した薄膜で 1 cm2V-1s-1前後と報告されています。
このため、C60そのものの電子輸送材料としての価値は非常に高いと言えます。
しかしながら、有機半導体デバイスを実用的なものにするためには、用いられている有機半導体材料が
「低コスト」「大面積」を可能にするデバイス作成プロセスに適応している必要があります。
真空蒸着ではこのような低コスト・大面積を実現することは難しいため、
有機半導体材料を溶媒に溶かして薄膜を作成する溶液プロセスが主に用いられます。
溶液プロセスを用いるためには材料を溶媒に溶解させる必要がありますが、
C60そのものの溶媒への溶解度は低いため、
一般的に、C60を化学修飾してC60誘導体として用います。
最も良く知られた例としては、
[6,6]-phenyl C61 butylic acid methyl ester(PCBM)といったものがあります。
このようなC60誘導体を電子輸送材料として用いる場合、
・C60とC60誘導体(A)の電子移動度は同じなのか?違うのか?
・C60誘導体(A)とC60誘導体(B)の電子移動度は同じなのか?違うのか?
という疑問が当然浮かびます。
そこで、我々(徳永、川畑)は化学修飾がC60の電子輸送特性に及ぼす影響について理論的な考察を行っています。
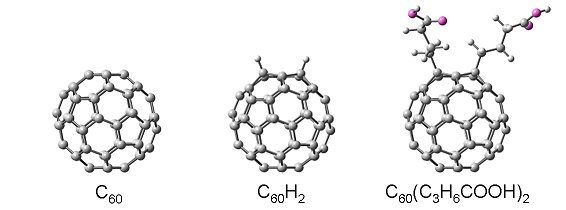
以下のように、C60と2種類のC60誘導体を考えます。
左に示すのはC60、
中央に示すのは最も簡単なC60誘導体であるC60H2、
そして右に示すのは-COOH末端を有するC60誘導体:C60(C3H6COOH)2です。
|
 |
我々の計算で、
・C60とC60誘導体(C60H2, C60(C3H6COOH)2)の電子輸送特性には非常に大きな違いがある
・しかし、C60H2とC60(C3H6COOH)2の電子輸送特性には殆ど差はない
ということが分かっています(本・雑誌 3、論文 15)。言いかえると、
・C60とC60誘導体を同じと考えるのはNG
・しかし、C60(C3H6COOH)2とC60H2を同じと考えるのはOK
ということになります。
なぜこのように考えても良いのかといいますと、
C60誘導体の電子移動特性を支配するのは、
化学修飾によるC60部位の電子構造・幾何学的構造の変化であるためです。
C60部位の電子構造・幾何学的構造の変化は、
「付加しているものの位置」に強く依存しており、
「付加しているものの種類(化学的性質)」は殆ど影響を及ぼしません。
ですので、付加しているものを水素原子で簡略化しても良く、そうして議論するのが一番簡単、ということになります。
以上の結果は様々なC60誘導体に適応できると考えられるので、
C60誘導体を用いた材料設計において、非常に有用な知見になるものと期待しています。
この結果に基づき、C60X4, C60X6 (投稿中)の研究を進めています。
この他、C60(CF2)n(論文 14)タイプのC60誘導体、
C70H1(論文 20),
C70H2(論文 18)といったC70誘導体の研究も進めています。
|
| |
| |
| 4. 振電相互作用定数の評価・解析と化学反応への応用 |
| (※)記述を簡潔にするため、表現が厳密ではない部分があります。詳細は論文をご覧下さい。 |
| |
| 振電相互作用(Vibronic Coupling)とは、文字通り
「振動状態(核の運動)」と「電子状態(電子の運動)」の相互作用のことを言います。
この相互作用は、一般的に |
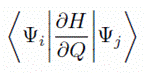 |
といった積分で表されます。
ここで、Ψ は全電子波動関数を、H はハミルトニアンを、Q はある振動モードの基準座標を表しています。
i = j の項は、分子に働く「力」を表します。例えば、ある分子をイオン化したときに、
その分子がある振動モードに沿ってどの程度変形するか?、ということなどを議論できます。
また、i ≠ j の項は、異なる電子状態間の分子振動を介した相互作用を表します。
この例として、「gerade → gerade への遷移は電子的禁制遷移であるはずなのに、弱いながらも遷移強度を有する」、いわゆる、
Intensity borrowing といったものが挙げられます。
これは、振動によって gerade の状態が ungerade の状態と混ざってるため、と解釈できます。
私のこれまで・これからの研究は、主に i = j の項を計算・解析することに重点を置いています。
以後、この積分値を振電相互作用定数(VCC)と呼ぶことにします。
当然のことながら、この VCC の計算・解析は 50 年以上前から興味が持たれていたのですが、
不思議なことに、計算方法・解析方法に統一性が見られていませんでした。
つまり、論文などで、VCC を計算した・解析したと書かれていても、
どこか「しっくり」来ない部分が残されていました。
具体例を示しますと、VCC の計算に関しては、
「VCC = エネルギー勾配 」としている論文があるが、これは正しいのか?
縮退点を一つの Slater 行列式に基づく計算方法で計算しても良いのか?
また、解析に関しては、「VCC の大きさ ⇔ フロンティア軌道の結合性」のように関係付けることは正しいのか?などです。
他にもありますが、ここでは省略させて頂きます。
これらの「しっくり」来ない部分を払拭するため、我々(佐藤、徳永、田中)は以下に示すような VCC の計算方法・解析方法を確立しました。
「縮退系(論文 3)」と「非縮退系(論文 7)」で取り扱いが異なるので、分けて書きます。
|
| 1.対象とする現象の例・一般的に用いる参照核配置(R) |
| |
縮退系 |
非縮退系 |
| 現象の例 |
ヤーン・テラー効果 |
電子状態の変化に伴う構造緩和 |
| 参照核配置(R) |
円錐交差点 |
電子状態の変化前の平衡核配置 |
| |
| 2.参照核配置における波動関数の計算 |
| |
縮退系 |
非縮退系 |
| 計算するのは |
核配置(R)で計算:1回だけ |
核配置(R)で状態の変化前・後:計2回 |
| 計算方法 |
状態の平均化が必要 * |
何でも良い |
| 基底関数 |
何でも良い |
1次導関数の追加が必要 ** |
| * 縮退波動関数の対称性の破れを防ぐため。 |
| ** Hellmann-Feynmanの定理を成立させるため。 |
| |
| 3.得られた波動関数を用いて VCC を計算するときは、 |
| |
縮退系 |
非縮退系 |
| |
定義に従い計算 *** |
定義に従い計算 *** |
| *** エネルギー勾配などを VCC とみなしてしまうと、次の4.に示す解析方法が使えなくなる。 |
| |
| 4.VCC の大きさを解析するときに注目するのは |
| |
縮退系 |
非縮退系 |
| ① 電子に関して |
フロンティア電子密度 **** |
状態変化前・後の差電子密度 |
| ② 振動に関して |
振動の及ぼすポテンシャル変化 |
振動の及ぼすポテンシャル変化 |
| |
① と ② の分布の一致から、VCCの大きさを議論できる |
| **** 近似の結果ではなく、Clebsch-Gordan 係数の対称性から導かれる厳密なものである。 |
| |
| 以上、VCC の計算・解析における重要なポイントは、 |
| ・まず、適切な方法で電子波動関数を計算する |
| ・そして、VCC をその定義に従い計算することで、解析が行いやすくなる |
| ・つまり、① と ② の分布の比較に基づく、振電相互作用の局所的描像から解析できる |
| ・この局所的描像(空間分布)を我々は「振電相互作用密度」と呼んでいます |
| |
| 今後は、「振電相互作用密度」を用いた解析を、
化学反応の局所的描像の記述に応用することを目的としています。 |
| |
| |
| |